Our group uses computer simulations to understand and control molecular scale driving forces for a wide range of applications spanning biotechnology to fuel combustion. We develop new methods that expand the capabilities of molecular simulation and use advanced research computing resources to solve challenging problems in the area of computational molecular science. A few representative projects are described below.
Molecular Data Science
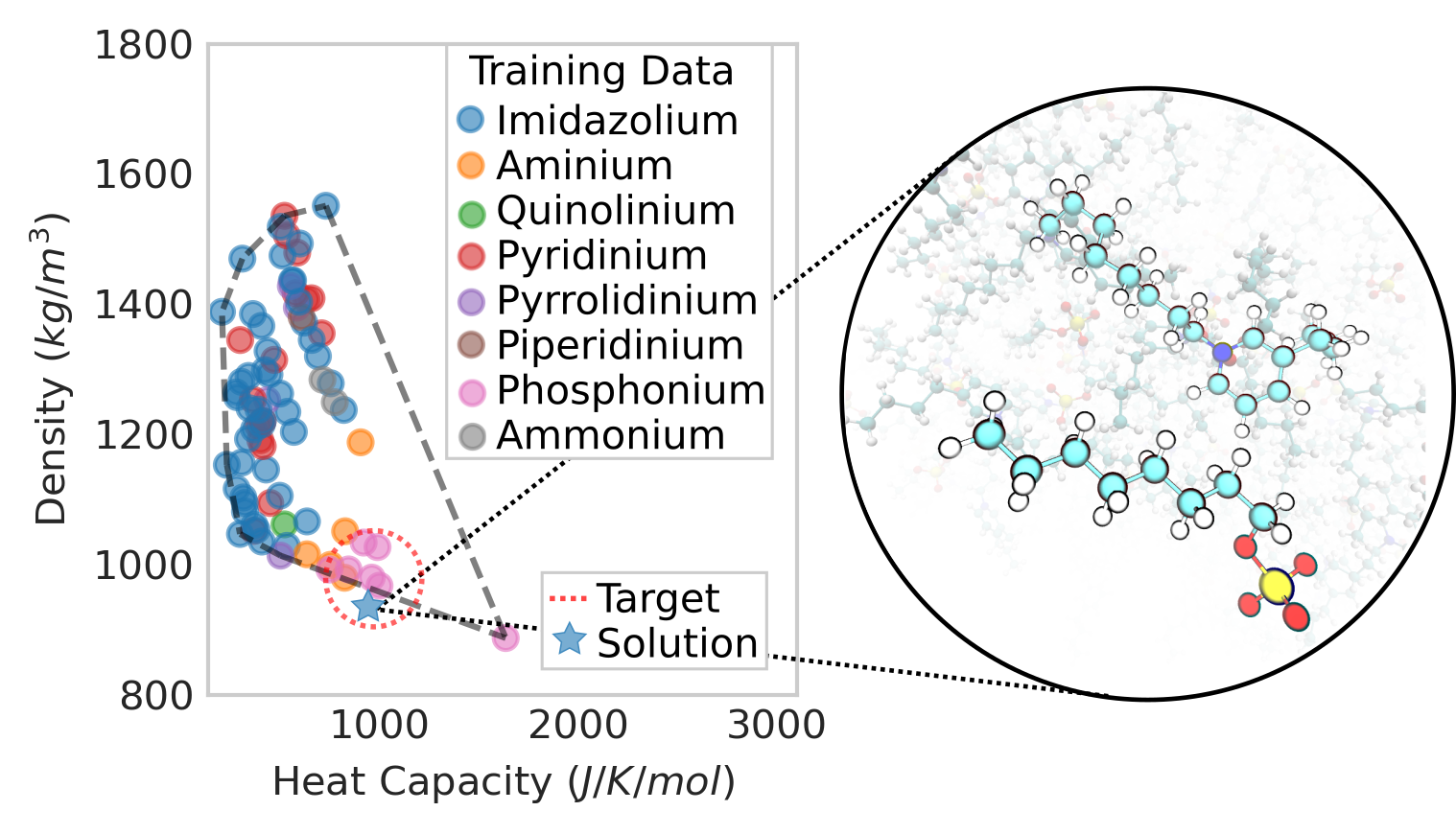
Molecular Data Science research in the Pfaendtner group involves using data visualization and statistical learning tools to aid in the discovery, design and characterization of new molecules and materials. Examples include development of surrogate models to aid in the rapid prediction of thermophysical properties or evolution of chemical reaction networks. Model systems where we apply these tools include protein-protein interactions, ionic liquids and biomass pyrolysis. We are actively developing a toolkit to drive the discovery of new molecules via genetic algorithm.
Recent Publications
- Wesley Beckner, Coco M. Mao, Jim Pfaendtner, "Statistical models are able to predict ionic liquid viscosity across a wide range of chemical functionalities and experimental conditions", Molecular Systems Design & Engineering, 2018, 3(1), 253-263. doi:10.1039/C7ME00094D
- Josh K. Smith, Kayla G. Sprenger, Rick Liao, Andrea Joseph, Elizabeth NanceJim Pfaendtner, "Determining dominant driving forces affecting controlled protein release from polymeric nanoparticles", Biointerphases 2017, 12(2), 02D412. doi:10.1116/1.4983154
- Blake R. Hough, Daniel T. Schwartz, Jim Pfaendtner, "Detailed kinetic modeling of lignin pyrolysis for process optimization", Industrial & Engineering Chemistry Research, 2016, 55(34), 9147-9153. doi:10.1021/acs.iecr.6b02092
Contributors
Biomolecular Simulations
Proteins at Interfaces
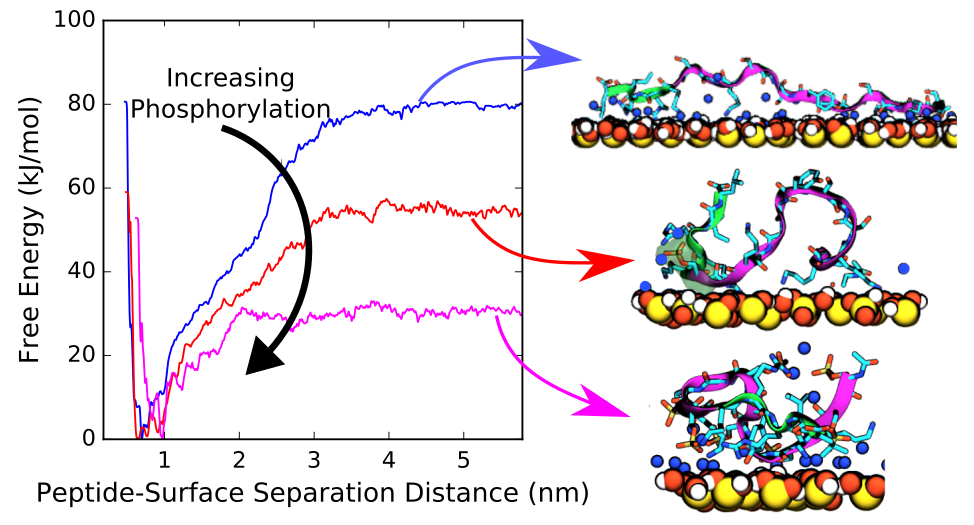
We use atomistic molecular simulations to study the behavior of biomolecules at interfaces with applications in biomaterials, biomineralization, and biotechnology. Examples include, peptide binding to silica surfaces to understand biomineralization or bio-inspired polymer self-assembly over mica. We use classical molecular dynamics to reveal the key molecular scale driving forces, but classical simulations are often insufficient to observe interfacial phenomena of biomolecules on the time and length scales accessible by even the most powerful supercomputers. Our group specializes in applying and developing enhanced sampling techniques like metadynamics to fully explore conformational space of these system, permitting estimates of relevant thermodynamic and kinetic quantities.
Proteins in Solution
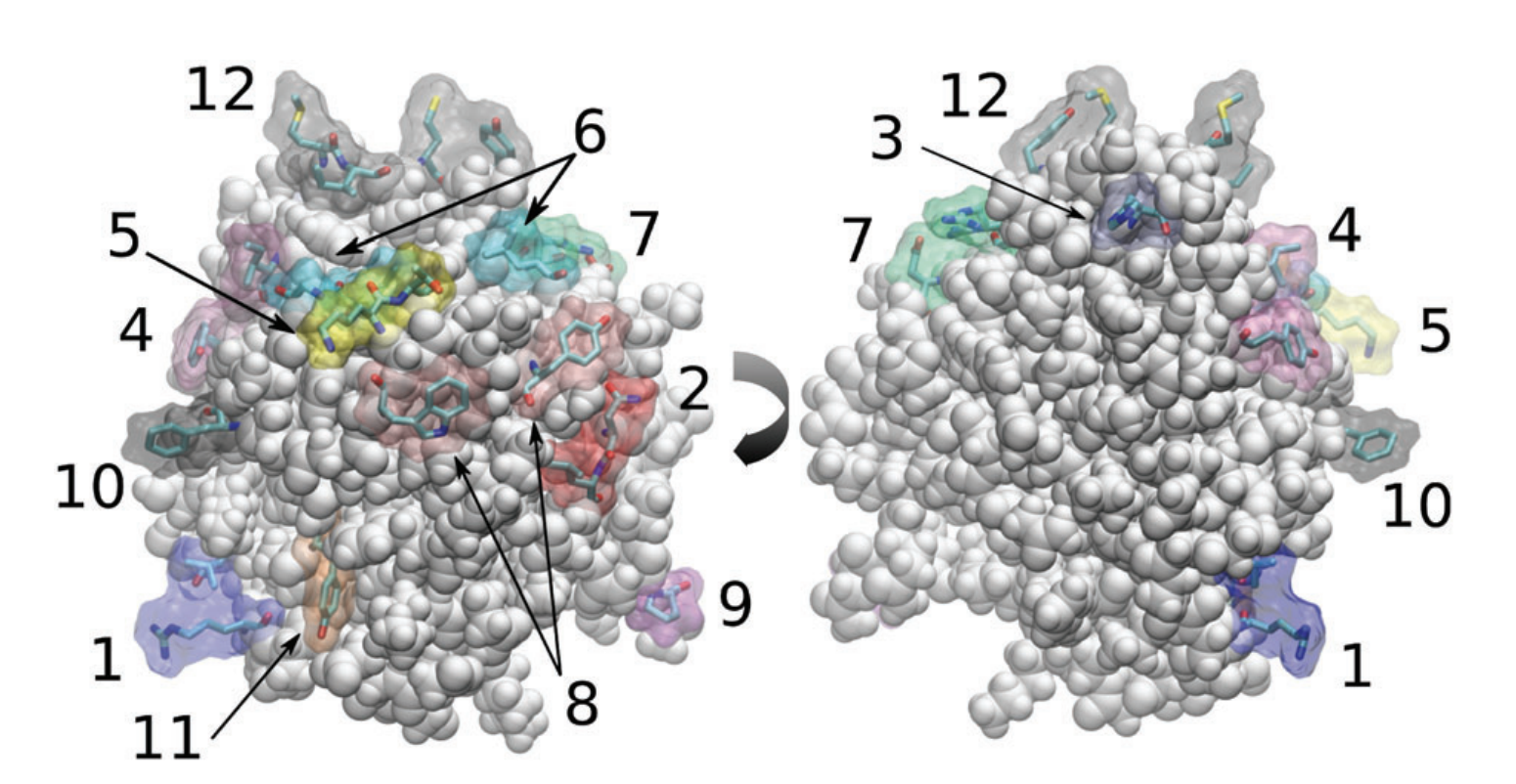
We also use MD simulations to study interactions between proteins and solvents. These simulations vary from studying the way proteins behave at the air water interface, to exploring how proteins interact in nonaqueous solvents. An example is using classical simulations to revealed the preferential binding patterns of IL solvents on carbohydrate active enzymes for applications in biocatalysis and biomass processing.
Recent Publications
- Arushi Prakash, K.G.Sprenger, Jim Pfaendtner, "Essential slow degrees of freedom in protein-surface simulations: A metadynamics investigation", BBRC, 2018, 498(2), 274-281. doi:10.1016/j.bbrc.2017.07.066
- Arushi Prakash, Marcel D Baer, Christopher J Mundy, Jim Pfaendtner, "Peptoid Backbone Flexibilility Dictates Its Interaction with Water and Surfaces: A Molecular Dynamics Investigation", Biomacromolecules, 2018, 19(3), 1006-1015. doi:10.1021/acs.biomac.7b01813
- K.G.Sprenger, J.G. Plaks, J.L. Kaar, Jim Pfaendtner, "Elucidating sequence and solvent specific design targets to protect and stabilize enzymes for biocatalysis in ionic liquids", PCCP, 2017, 19(26), 17426-17433. doi: 10.1039/C7CP03013D
Contributors
Reaction Networks
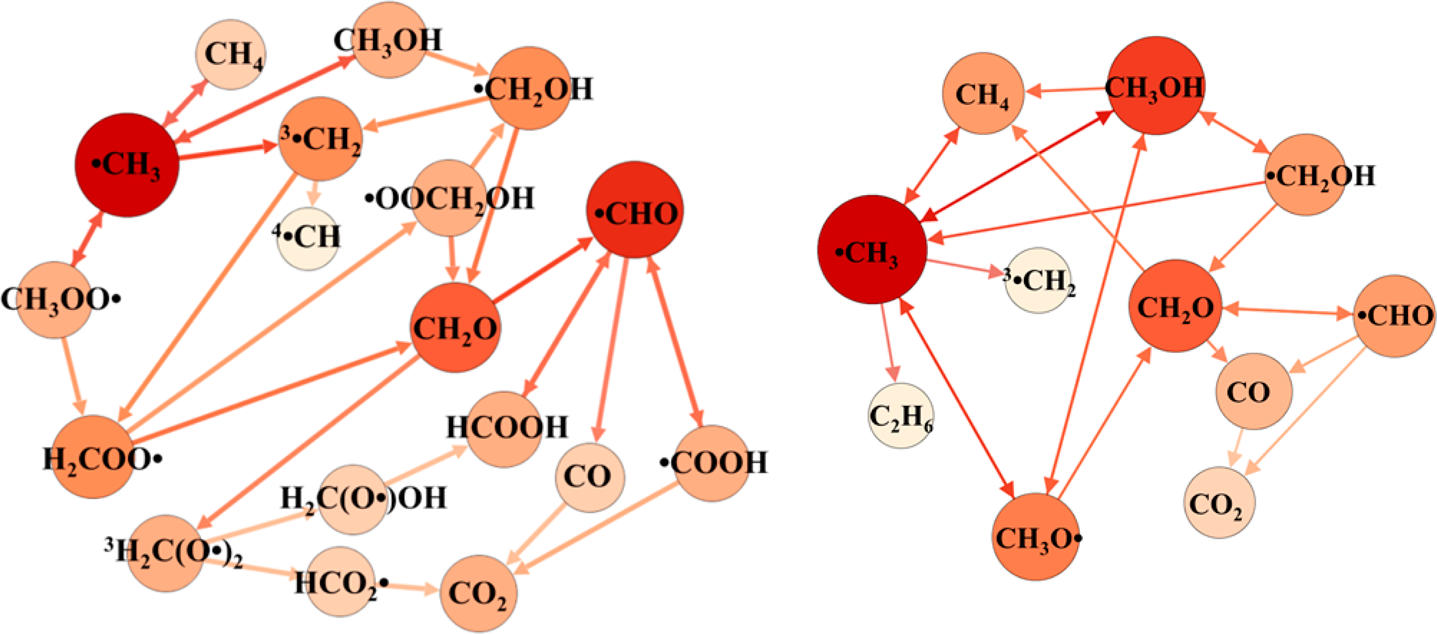
Our research in complex reaction networks primarily focuses on the use of MD simulation to predict new reaction pathways when the chemistry is too complicated to enumerate by hand and/or there is insufficient knowledge to apply automated reaction mechanism generation tools. We use a variety of approaches to characterize reaction networks including statistical learning, enhanced sampling, and ab initio methods. Current areas of interest include applications in combustion, pyrolysis and reactions in the condensed phase.
Recent Publications
- C. D. Fu, Jim Pfaendtner, "Lifting the Curse of Dimensionality on Enhanced Sampling of Reaction Networks with Parallel Bias Metadynamics", J. Chem. Theory Comput., 2018, 14(5), 2516–2525. doi: 10.1021/acs.jctc.7b01289
- Blake Hough, David A. C. Beck, Daniel T. Schwartz, Jim Pfaendtner, "Application of machine learning to pyrolysis reaction networks: Reducing model solution time to enable process optimization", Comput. Chem. Eng., 2017, 104, 56-63. doi: 10.1016/j.compchemeng.2017.04.012
- C. D. Fu, Luiz F. L. Oliveira, Jim Pfaendtner, "Assessing Generic Collective Variables for Determining Reaction Rates in Metadynamics Simulations", J. Chem. Theory Comput., 2017, 13(3), 968–973. doi: 10.1021/acs.jctc.7b00038
Contributors
Clean Energy Materials
We are using molecular simulations to study the evolution of concentrated electrolytes in the condensed phase and at interfaces. Both the condensed phase and interface present challenges for predicting the chemical reaction networks and characterizing the network through kinetic and thermodynamic analysis. Given the difficulties in probing chemical reactions interfaces with spectroscopic or other experimental approaches, computer simulations can play an enormous role in developing fundamental molecular understanding and creating predictive models. This project also develops new computational approaches that address time scale limitations with a suite of enhanced sampling tools such as infrequent metadynamics and high dimensional sampling schemes. Other areas of interest in the clean energy space include the design of custom molecules for applications such as solar collectors or flow batteries and the characterization of fast pyrolysis systems.
Recent Publications
- Blake Hough, Daniel T. Schwartz, Jim Pfaendtner, "Detailed Kinetic Modeling of Lignin Pyrolysis for Process Optimization", 2016, 55 (34), 9147-9153. doi: 10.1021/acs.iecr.6b02092